



The brain slices underwent directional cooling via a copper cylinder immersed in liquid nitrogen. (Image Credit: bioRxiv)
For the first time, scientists at the University of Erlangen-Nuremberg cryogenically froze and revived hippocampal slices of mouse brain tissue without harming them. The team used vitrification, subjecting the slices to cryoprotectants to keep them safe from ice crystals.
First, the team collected adult mouse hippocampal slice samples and placed them on a polyester mesh. Each sample was also subjected to rapidly increasing levels of cryoprotectant fluid at 50°F, then at 14°F.
Afterward, the team used a cylinder of liquid nitrogen to cool the slices directionally. They stayed in that spot for one minute and were carefully submerged in the frozen liquid. Using this gentle approach ensured the samples would not undergo physical cracking due to thermomechanical stress. The brain slices cooled to -321°F, causing the brain tissue water to turn into vitreous, non-crystalline material. Keeping them safe from ice crystal formation was achieved through an optimized vitrification process with cryoprotectants called V3. Before developing V3, the team tested varying mixtures of cryoprotectant agents.
V3 has a combination of dimethyl sulfoxide, formamide, and ethylene glycol. In this case, the cryoprotectants are nontoxic and reduce tissue damage due to swelling, cracking, crystallization, and shrinking.
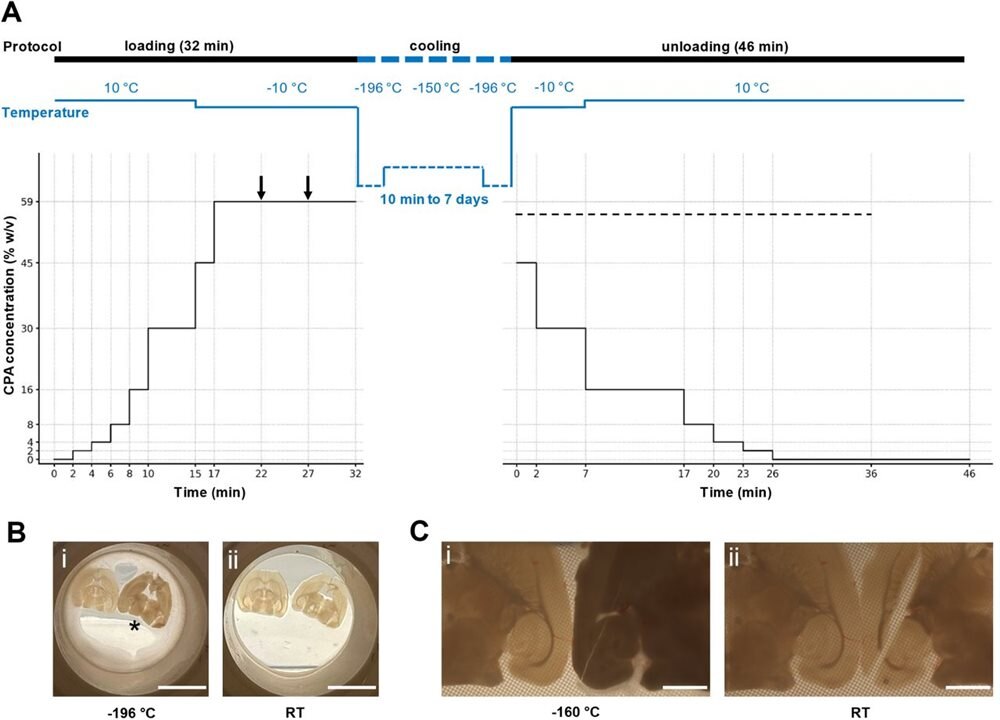
The team created an optimized vitrification process involving temperature and cryoprotectant concentration changes. (Image Credit: bioRxiv)
The researchers placed the brain tissues in a freezer (-238°F) for a week. They then carefully thawed each sample by removing them from the freezer and placing them in liquid nitrogen. After that, they put the samples back on the cylinder top and rewarmed the slices in V3 at 14°F to wake them up. They also kept osmotic shock from settling in by reducing the V3 cryoprotectant, which was substituted with artificial cerebrospinal fluid (aCSF) with mannitol. Each slice was then maintained at room temperature in carbogenated aCSF for two hours for recovery.
The team used a Leica Ivesta 3 video stereomicroscope to inspect the brain slices. According to the paper, “for light microscopy to evaluate brain structure and neural morphology, the brain slices with standard CPA loading of 59% w/v, with and without vitrification, were fixated with 4% paraformaldehyde overnight at 4 °C, washed, and kept in phosphate-buffered saline (PBS).”
For electron microscopy analysis, the team prepared each vitrified slice through several steps. The samples were fixed in a solution of 2% glutaraldehyde and 1% formaldehyde cacodylate buffer for 24 hours. Afterward, they washed them overnight in cacodylate buffer and post-fixed with osmium tetroxide (OsO4). Once dehydrated, the team placed the tissue in Epon resin. Semithin (1 μm thick) and ultrathin sections (50 nm thick) were formed. These were stained with uranyl acetate and lead citrate. Lastly, the team observed the ultrathin sections using a Zeiss EM 800N electron microscope, providing a detailed view of the tissue’s ultrastructure.
The team observed the tissues and found they had no crystallization while being cooled or rewarmed. After revival, the samples exhibited near-normal electrical activity and maintained neural functionality. Using electrophysiology equipment, the team detected all the responsive synapses connecting neurons that remained intact. It's also possible that memories could be preserved as the team observed long-term potentiation, a key neurobiological substrate of learning and memory formation. This makes it possible to place the brain in suspended animation and continue its activity.
Have a story tip? Message me at: http://twitter.com/Cabe_Atwell